Amyloidosis
Amyloidosis is the deposition of an abnormal substance called amyloid in the tissues of the body. Amyloidosis is not a disease as much as it is a specific biochemical arrangement in which a precursor protein or protein fragment accumulates and forms filaments. These filaments align and stack themselves in a beta-sheet conformation forming fibrils that twist around each other and are deposited in the tissues as amyloid deposits. Amyloid deposits differ due to the nature of the protein precursors that form there. Due to this structure amyloid is insoluble and can be thought of as “wax”. There are currently at least 11 different protein precursors in amyloidosis. Many of these have specific tissues they accumulate in and have specific clinical signs or syndromes associated with them. As an example in humans Alzheimer’s patients have amyloid deposition in the brain due to an unknown precursor protein. In animals the only form of amyloidosis shown to occur at this time is reactive amyloidosis. Here the precursor protein is serum amyloid A protein and amyloid A composes the fibrils in the amyloid deposits. All amyloid deposits stain with Congo Red and have a characteristic green color with birefringence under polarized light. Of the many types of amyloidosis which stain with Congo Red only reactive amyloid and beta-2 microglobulin amyloid are decolorized by potassium permanganate oxidation. Beta-2 microglobulin amyloid has only been described in humans on long-term hemodialysis for chronic renal failure and has not been found in animals. The amyloid in Shar-Pei is definitively reactive amyloidosis. It is also important to realize that amyloidosis is not a single disease, but can be the end point of many diseases.
This form of systemic amyloidosis, reactive amyloidosis, usually occurs with chronic inflammatory diseases and is characterized by the presence of amyloid protein AA. Amyloid protein AA is derived from an acute phase protein called serum amyloid A protein (SAA) produced by the liver. There are many other acute phase proteins produced by the liver which have important roles in the inflammatory process and in tissue repair after injury. It is important to understand that amyloid protein AA is a normal protein and that it’s production is a normal response to tissue injury and inflammation. It is also important to realize that many diseases, traumatic injuries, cancer disorders, stresses, etc. can stimulate the production of the acute phase proteins. There appears to be a balance between the production of SAA and the degradation and excretion of SAA from the body. It is not known whether the development of amyloidosis in the Shar-Pei is due to prolonged excessive SAA production by the liver which overwhelms the degradation mechanisms or a defect in the degradation process itself, or a combination of both. We do know that Familial Shar-Pei Fever is an inflammatory process which does stimulate the synthesis and release of acute phase proteins from the liver. That this occurs can be surmised from the changes seen on the hemogram and biochemical profiles of Shar-Pei during, or shortly after, an FSF episode. It certainly appears that the cause of amyloidosis in Shar-Pei has a genetic basis.
Reactive amyloidosis results in extracellular deposition of amyloid protein in tissues. This means the “waxy” amyloid is surrounding the cells and slowly crushes them as well as interfering with nutrition of the cells. These cells die and the structures they make up are replaced by fibrous, nonfunctional scar tissue. There are species differences as to which tissues amyloid will accumulate in. In dogs, the kidney is the primary organ involved with the spleen and liver affected less often. The kidney is especially vulnerable due to its decreased ability to replace damaged cells and ultimately, when a certain number of cells have been irreparably damaged, kidney failure with its accompanying clinical signs develops. Once amyloid is deposited in the tissues it appears that nothing can remove it although recently a new class of drugs have been discovered which may do just that.
It is important to realize that amyloidosis occurs in all dog breeds and in a number of inflammatory and immune-mediated conditions. The majority of amyloidosis in dogs is idiopathic or of unknown cause. Amyloidosis can occur secondary to heartworm disease, tick-borne diseases, various cancers, systemic lupus, immune-mediated arthritis, IBD, etc. I suspect that the tendency to develop amyloidosis in response to inflammatory or immune-mediated disease or triggers is a separate genetic disease in dogs which leads to failure to degrade amyloid, failure to excrete amyloid degradation products or increased production of amyloid precursors.
Why does amyloidosis have so many different clinical presentations? Why does it occur in some Shar-Pei at 2 years of age and in others at 10 years of age? Why do some Shar-Pei develop amyloidosis and others don’t? Why is it a genetic disease in Shar-Pei? There are many questions which have no answers at this time. I think several theories are plausible to explain the variations we see:
The underlying trigger of amyloidosis in Shar-Pei is Familial Shar-Pei Fever (FSF). It is quite possible that FSF has variable age of onset and variable degrees of severity in terms of the inflammatory disease it causes. This may result in a variable rate of progression in the development of amyloidosis in different individuals. For example, the response of the liver to FSF and the synthesis and release of the acute phase proteins, especially SAA, may be more acute in some dogs resulting in a more rapid deposition of amyloid. In other individuals, the response to FSF may be more chromic and result in slower deposition of amyloid. In effect, there may be milder forms and more severe forms of the same disease. The exact mechanism of amyloid deposition may be different in different individuals. There may be other effects of FSF on the body which are additive with the amyloidosis. As an example, we know Shar-Pei are more susceptible to disseminated intravascular coagulation (internal blood clotting) during an episode of FSF and blood clots in the kidneys may cause more kidney damage than just amyloid deposition itself. Some dogs may have other disease processes going on which can be additive with the effects of amyloidosis.
These are just some ideas on why we see different presentations of the same disease. One fact remains – any amyloid deposits found in a Shar-Pei have to be regarded as related to FSF and genetic until proven otherwise. It really doesn’t matter whether a little or a large amount of amyloid is found. Another point to keep in mind is that the mechanisms initiating amyloid deposition are normal protective responses seen in any breed of dog. It appears in our breed that the mechanisms, which regulate the inflammatory response, don’t work properly allowing this normal response to go out of control and cause disease.
Relationship of FSF and Amyloidosis
There is a definite relationship between FSF and amyloidosis. FSF is undoubtedly a genetic condition which is inherited much as the periodic fever episodes seen in humans. FSF is a built-in trigger resulting in intense inflammation and the release of acute phase proteins from the liver one of which is serum amyloid A. I think there are several explanations for the wide variety of clinical presentations we see with FSF/amyloidosis. I suspect that there are several different mutations which can result in FSF. We know in humans with Familial Mediterranean Fever that there are at least 8 mutations in the FMF gene leading to differences in severity of clinical signs, duration, illness and death in different populations. I think we see the same thing in Shar-Pei, some having mild episodes and some having very severe episodes. This may also explain why some dogs succumb to STSS, DIC, heat stress, and other complications but others don’t. That being said I also suspect that there are variations in the genetic defect predisposing to amyloidosis in dogs in general. One can imagine in a dog with a severe form of FSF resulting in dramatic increases in serum amyloid A and having a genetic defect leading to a decrease in degradation of amyloid that the rate of amyloid deposition would be very high. On the other hand if the type of FSF resulted in mild increases in serum amyloid A in a dog with no amyloid defect then amyloidosis would not develop. The same seems to hold true with the dog population in general since not all dogs with inflammatory/immune-mediated diseases develop amyloidosis and some dogs with amyloidosis have no discernable underlying disease to blame for it.
I think it is safe to say that FSF is not the only precursor to the development of amyloidosis in the Shar-Pei but certainly is the major factor since FSF is only seen in our breed. Is it possible that FSF is revealing those animals with an amyloidosis defect of some kind? The high fever seen in FSF certainly results in increased levels of serum amyloid A thus facilitating the development of amyloidosis. Thus it appears FSF and amyloidosis most often occur together in Shar-Pei but can occur separately as well. Since idiopathic amyloidosis occurs in dogs in general (the most common cause of amyloidosis in other breeds) we can expect it would occur in the Shar-Pei also so not all cases of amyloidosis in our breed are related to FSF.
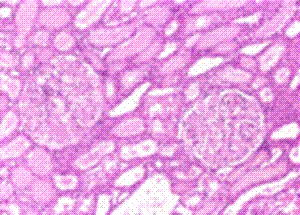
H & E Stain: The pink-staining material in the collecting ducts, tubules and glomeruli is amyloid. Unfortunately protein in the tubules will also appear the same. Only Congo Red staining can differentiate amyloid deposits.
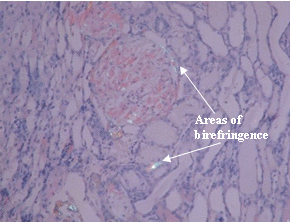
Congo Red Stain: The areas of birefringence are due to amyloid.